
案例介绍
没有,但是在做md模拟。
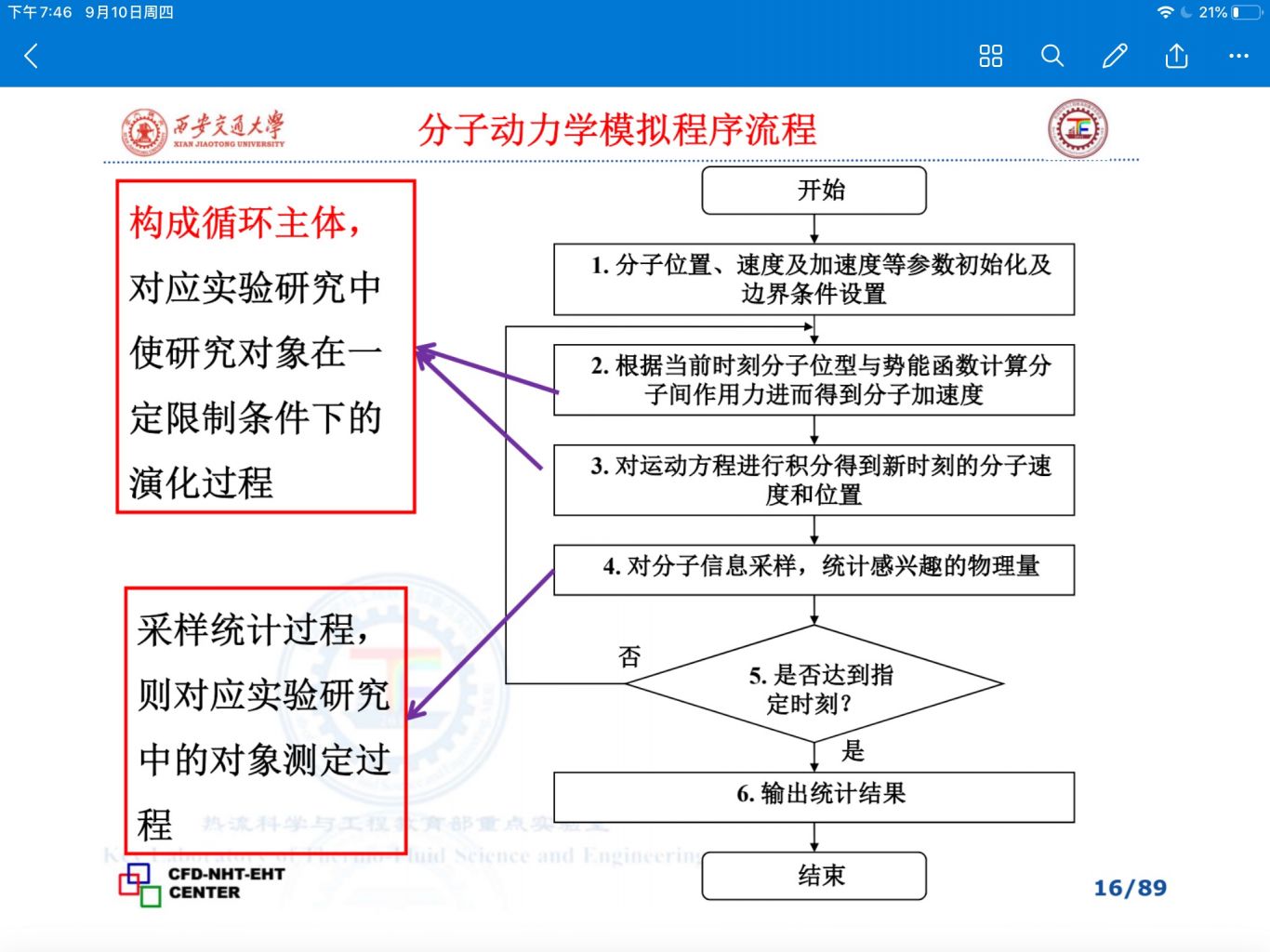
分子动力学是一门结合物理,数学和化学的综合技术。分子动力学是一套分子模拟方法,该方法主要是依靠牛顿力学来模拟分子体系的运动,以在由分子体系的不同状态构成的系综中抽取样本,从而计算体系的构型积分,并以构型积分的结果为基础进一步计算体系的热力学量和其他宏观性质。
基本步骤
确定起始构型
进行分子动力学模拟的第一步是确定起始构型, 一个能量较低的起始 构型 是进行 分 子模拟 的基础 ,一般分子的起始构型主要来自实验数据或量子化学计算。
在确定起始构型之后要赋予构成分子的各个原子速度,这一速度是根据波尔兹曼分布随机生成的,由于速度的分布符合波尔兹曼统计,因此在这个阶段,体系的温度是恒定的。另外,在随机生成各个原子的运动速度之后须 进行调整,使得体系总体在各个方向上的动量之和为零,即保证体系没有平动位移。
进入平衡相
由上 一步 确定的分子组建平衡相,在构建平衡相的时候会对构型、温度等参数加以监控。
进入生产相
进入生产相之后体系中的分子和分子中的原子开始根据初始速度运动,可以想象其间会发生吸引、排斥乃至碰撞,这时就根据牛顿力学和预先给定的粒子间相互作用势来对各个粒子的运动轨迹进行 计算,在这个过程中,体系总能量不变,但分子内部势能和动能不断相互转化,从而 体系的温度也不断变化,在整个过程中,体系会遍历势能面上的各个点,计算的样本正是在这个过程中抽取的。 +
计算结果
案例图片

相似案例推荐
其他人才的相似案例推荐
-

鸿儒专升本
大小自考,教师资格证,会计等考试提供视频和试题资料。通过后台
-
")
无痕养育成长营小程序(会员版)
作品简介:在线教育类小程序,尹建莉父母学堂公众号拥有超过50
-

西安电子科技大学基金会系统
西安电子科技大学基金会系统为管理学校各项赞助资金及设立基金管
-

西安电子科技大学校友会/基金会系统
西安电子科技大学校友会系统为收集毕业生及在校生基本信息,使毕
-

在线教育平台
在线付费视频直播 and 聊天室 and 论坛 a
-

四川自考网
网站内容是客户提供进行相应的制作 是通过phpcms技术进行
-

教务管理系统
系统详细功能: 班级信息管理: 本模块详细的录入了全校各个
-

作业评管教师端
作业评管教师端,学生端通过与后台配合使用,后台发布作业或考试
-

知夫家教
此app的主要目的是为了方便大学生找家教,做的一个一个资源整
-

创社服盟
此图片未创社服盟的部分页面,具体不便透露,详细请联系时再详细
-
")
南京市科利华中学(微信推送板块)
使用PHP定制接口。 缓存及懒加载技术。 同“微校+”平
-

教育统一网上办事大厅
作品主要为了面向政务教育行业的项目,当然提供B2C。主要做用
微信接收人才推送
关注猿急送微信平台,接收实时人才推送

